Hierarchical Clustering Analysis - MASE#
In this notebook, we will use multiple adjacency spectral embedding (MASE) instead of omnibus embedding to obtain a low dimensional vector representation of vertices in our correlation matrices
import pandas as pd
import graspologic as gp
import seaborn as sns
import hyppo
import matplotlib.pyplot as plt
import numpy as np
import plotly.graph_objects as go
import matplotlib.cm
from sklearn.preprocessing import LabelEncoder
%matplotlib inline
import plotly.io as pio
pio.renderers.default="png"
# ## Read the brain region information
# t = pd.read_excel("../data/raw/CHASSSYMM3AtlasLegends.xlsx", engine='openpyxl')
# pd.concat([t, cluster_label_df], axis=1).to_csv("CHASSSYMM3AtlasLegends.csv", index=False)
# out = pd.concat([node_labels, cluster_label_df], axis=1)
# out.to_csv("./out.csv")
## Read the data
key = pd.read_csv('../data/processed/key.csv')
data = pd.read_csv('../data/processed/mouses-volumes.csv')
data.set_index(key.DWI, inplace=True)
genotypes = ['APOE22', 'APOE33', 'APOE44']
gen_animals = {genotype: None for genotype in genotypes}
for genotype in genotypes:
gen_animals[genotype] = key.loc[key['Genotype'] == genotype]['DWI'].tolist()
vol_dat = {genotype: [] for genotype in genotypes}
for genotype in genotypes:
vol_dat[genotype] = data.loc[gen_animals[genotype]].to_numpy()
## Compute correlations
cor_dat = {genotype: gp.utils.symmetrize(np.corrcoef(dat, rowvar=False)) for (genotype, dat) in vol_dat.items()}
## Node hierarchical label data
node_labels = pd.read_csv("../data/processed/node_label_dictionary.csv")
node_labels
| Structure | Abbreviation | Hemisphere | Level_1 | Level_2 | Level_3 | Level_4 | Subdivisions_7 | index | index2 | clusfound | Subdivisions_7_nowm | Hemisphere_abbrev | Level_1_abbrev | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | Cingulate_Cortex_Area_24a | A24a | Left | 1_forebrain | 1_secondary_prosencephalon | 1_isocortex | 2_cingulate_cortex | 1_isocortex | 1 | 1.0 | 0 | 1_isocortex | L | FB |
| 1 | Cingulate_Cortex_Area_24a_prime | A24aPrime | Left | 1_forebrain | 1_secondary_prosencephalon | 1_isocortex | 2_cingulate_cortex | 1_isocortex | 2 | 2.0 | 0 | 1_isocortex | L | FB |
| 2 | Cingulate_Cortex_Area_24b | A24b | Left | 1_forebrain | 1_secondary_prosencephalon | 1_isocortex | 2_cingulate_cortex | 1_isocortex | 3 | 3.0 | 0 | 1_isocortex | L | FB |
| 3 | Cingulate_Cortex_Area_24b_prime | A24bPrime | Left | 1_forebrain | 1_secondary_prosencephalon | 1_isocortex | 2_cingulate_cortex | 1_isocortex | 4 | 4.0 | 0 | 1_isocortex | L | FB |
| 4 | Cingulate_Cortex_Area_29a | A29a | Left | 1_forebrain | 1_secondary_prosencephalon | 1_isocortex | 2_cingulate_cortex | 1_isocortex | 5 | 5.0 | 0 | 1_isocortex | L | FB |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 327 | IntermediateReticularNucleus | IRN.1 | Right | 3_hindbrain | 2_pontine_hindbrain | NaN | axial_hindbrain_white_matter | 6_hindbrain | 328 | 1162.0 | 0 | 6_hindbrain | R | HB |
| 328 | PosteriorDorsal_ParaventricularMedialParvicell... | PHD_PaMP_Post_and_LatHy.1 | Right | 1_forebrain | 1_secondary_prosencephalon | 8_rostral_secondary_prosencephalon | 5_hypothalamus | 4_diencephalon | 329 | 1163.0 | 0 | 4_diencephalon | R | FB |
| 329 | Prerubral_Forel | PR_p3Tg_L.1 | Right | 1_forebrain | 2_diencephalon | p2_and_p3 | 17_thalamus | 4_diencephalon | 330 | 1164.0 | 0 | 4_diencephalon | R | FB |
| 330 | PVG_of_Hypothalamus | PVG.1 | Right | 1_forebrain | 1_secondary_prosencephalon | 8_rostral_secondary_prosencephalon | 5_hypothalamus | 4_diencephalon | 331 | 1165.0 | 0 | 4_diencephalon | R | FB |
| 331 | Basal Lateral Amygdala | BLA.1 | Right | 1_forebrain | 1_secondary_prosencephalon | 6_subpallium | 8_pallial_and_subpallial_amygdala | 2_pallium | 332 | 1166.0 | 0 | 2_pallium | R | FB |
332 rows × 14 columns
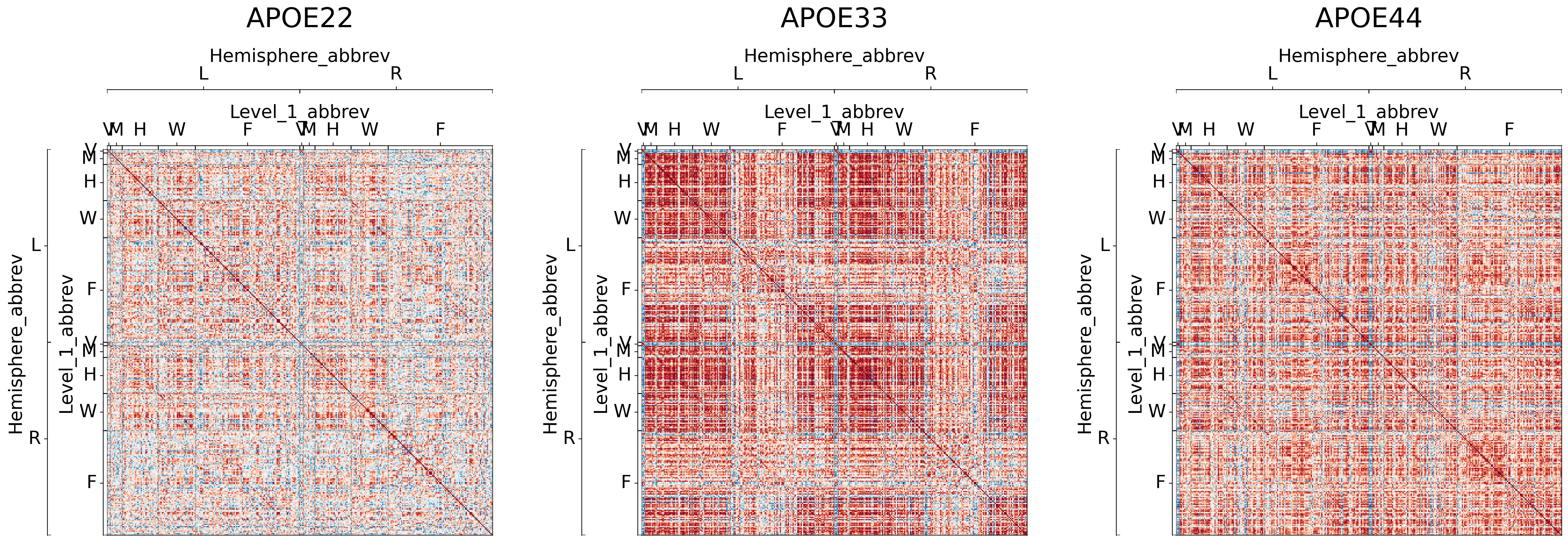
## Plot to make sure nothing is wrong
fig, ax = plt.subplots(
ncols=3,
figsize=(30, 10),
#constrained_layout=True,
dpi=300,
gridspec_kw=dict(width_ratios=[1, 1, 1])
)
for (i, genotype) in enumerate(cor_dat.keys()):
gp.plot.adjplot(
cor_dat[genotype],
ax=ax[i],
vmin=-1,
vmax=1,
meta=node_labels,
group=['Hemisphere_abbrev', 'Level_1_abbrev'],
)
ax[i].set_title(f"{genotype}", pad=135, size=30)
#fig.tight_layout(pad=3.0)
Embed the data simultaneously using Multiple Adjacency Spectral Embedding (MASE)#
The purpose of MASE is to obtain a low dimensional representation of the three correlation matrices such that the embedded correlation matrices can be compared to each other in a meaningful way [Arroyo et al., 2021]. Unlike omnibus embedding, MASE provides a common vertex representation across all correlation matrices. One can view this as a factoring out of common structures that are shared across all correlation matrices. This common representation of vertices provide a low dimensional vector per region of the brain, resulting in a \(322\times d\) matrix per genotype where \(d\) is the “embedding dimension” and \(d << 332\).
We will use the embeddings in order to perform hierchical clustering [Athey et al., 2019, Lyzinski et al., 2014, Priebe et al., 2019].
mase = gp.embed.MultipleASE()
Vhat = mase.fit_transform([corr for _, corr in cor_dat.items()])
Xhats = []
for score in mase.scores_:
u, d, v = np.linalg.svd(score)
Xhat = Vhat @ u @ np.diag(np.sqrt(d))
Xhats.append(Xhat)
Perform hierarchical clustering#
First we concatenate the embeddings resulting in a matrix with size \(332 \times 3d\). We will iteratively divide the regions into two clusters. For example, we divide the \(332\) regions into two clusters, and each resulting cluster will be divided into two more clusters, etc. The clustering algorithm used at each division is Gaussian mixture modeling.
The clustering algorithm will tell us which regions should be grouped together, and subsequently forms our “subgraphs” given by the clusterings.
Vhat.shape
(332, 2)
cluster = gp.cluster.DivisiveCluster(max_level=3)
cluster_labels = cluster.fit_predict(Vhat, fcluster=True)
cluster_label_df = pd.DataFrame(cluster_labels, columns=["cluster_level_1", "cluster_level_2", "cluster_level_3"])
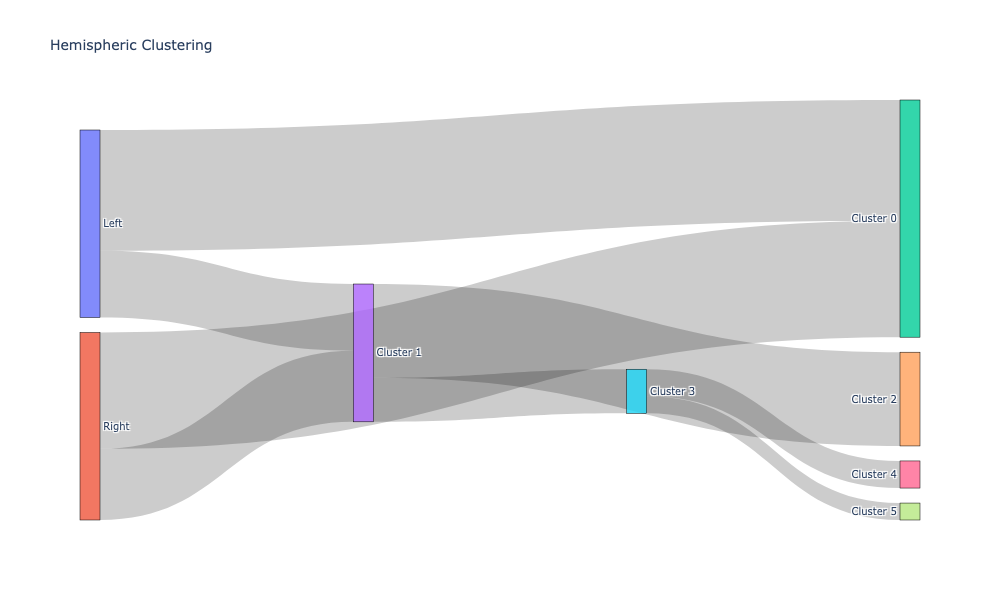
Plotting the cluster labeling along with apriori labels using Sankey diagrams#
Sankey diagrams tell us which regions or subregions belong to which clusters.
def count_groups(label_matrix):
levels = label_matrix.shape[1] - 1
d = []
for level in range(levels):
upper_cluster_ids = np.unique(label_matrix[:, level])
for upper_cluster_id in upper_cluster_ids:
lower_cluster_ids, counts = np.unique(
label_matrix[label_matrix[:, level] == upper_cluster_id][:, level + 1], return_counts=True
)
for idx, lower_cluster_id in enumerate(lower_cluster_ids):
if upper_cluster_id == lower_cluster_id:
lower_cluster_id = None
d.append((upper_cluster_id, lower_cluster_id, counts[idx]))
d = np.array(d)
source = d[:, 0]
target = d[:, 1]
value = d[:, 2]
return source, target, value
def append_apriori_labels(apriori_labels, cluster_matrix):
encoder = LabelEncoder()
apriori_labels_encoded = encoder.fit_transform(apriori_labels)
apriori_labels_encoded = apriori_labels_encoded.reshape(-1, 1)
# Increase the original cluster_matrix labels
cluster_matrix_ = cluster_matrix + np.max(apriori_labels_encoded) + 1
out = np.hstack([apriori_labels_encoded, cluster_matrix_])
return out, list(encoder.classes_)
hemispheric_clusters, encoded_labels = append_apriori_labels(node_labels.Hemisphere, cluster_labels)
source, target, value = count_groups(hemispheric_clusters)
fig = go.Figure(data=[go.Sankey(
node = dict(
pad = 15,
thickness = 20,
line = dict(color = "black", width = 0.5),
label = encoded_labels + [f"Cluster {i}" for i in range(np.max(hemispheric_clusters))]
),
link = dict(
source = source,
target = target,
value = value
))])
fig.update_layout(title_text="Hemispheric Clustering", font_size=10)
fig.show(dpi=300, width=1000, height=600)
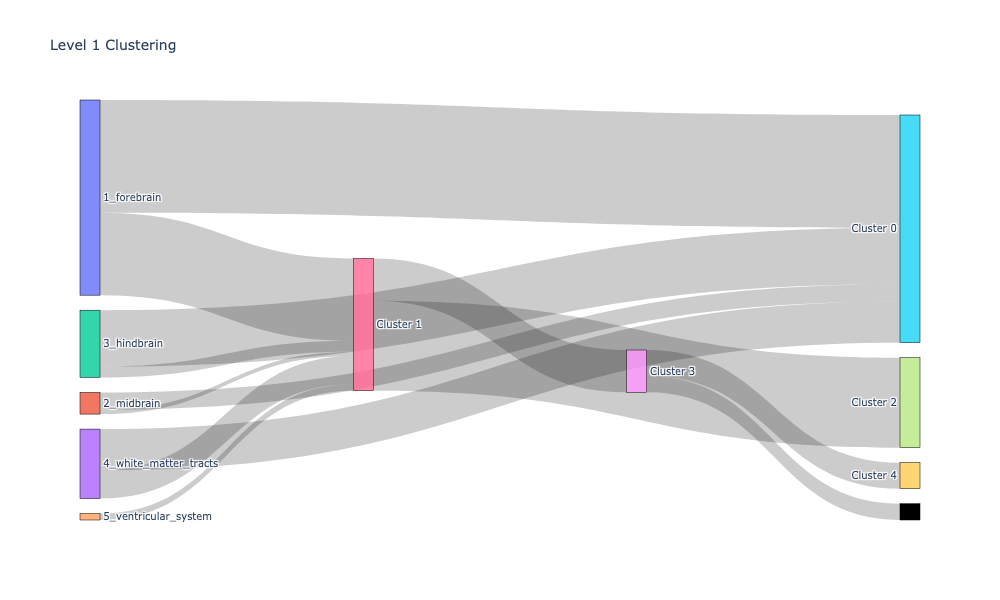
level_1_clusters, encoded_labels = append_apriori_labels(node_labels.Level_1, cluster_labels)
source, target, value = count_groups(level_1_clusters)
fig = go.Figure(data=[go.Sankey(
node = dict(
pad = 15,
thickness = 20,
line = dict(color = "black", width = 0.5),
label = encoded_labels + [f"Cluster {i}" for i in range(np.max(level_1_clusters[0]))],
),
link = dict(
source = source,
target = target,
value = value
))])
fig.update_layout(title_text="Level 1 Clustering", font_size=10)
fig.show(dpi=300, width=1000, height=600)
pd.concat([node_labels, cluster_label_df], axis=1).to_csv("../outs/mase_clustering.csv", index=False)
Visualizing different clustering levels using heatmaps#
Heatmaps can qualitatively tell us if there are any underlying structures within the clusters.
cl = pd.DataFrame(cluster_labels, columns=[f"Cluster_{i}" for i in range(1, 4)])
meta = pd.concat([node_labels, cl], axis=1)
meta.head()
| Structure | Abbreviation | Hemisphere | Level_1 | Level_2 | Level_3 | Level_4 | Subdivisions_7 | index | index2 | clusfound | Subdivisions_7_nowm | Hemisphere_abbrev | Level_1_abbrev | Cluster_1 | Cluster_2 | Cluster_3 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | Cingulate_Cortex_Area_24a | A24a | Left | 1_forebrain | 1_secondary_prosencephalon | 1_isocortex | 2_cingulate_cortex | 1_isocortex | 1 | 1.0 | 0 | 1_isocortex | L | F | 0 | 0 | 0 |
| 1 | Cingulate_Cortex_Area_24a_prime | A24aPrime | Left | 1_forebrain | 1_secondary_prosencephalon | 1_isocortex | 2_cingulate_cortex | 1_isocortex | 2 | 2.0 | 0 | 1_isocortex | L | F | 1 | 2 | 2 |
| 2 | Cingulate_Cortex_Area_24b | A24b | Left | 1_forebrain | 1_secondary_prosencephalon | 1_isocortex | 2_cingulate_cortex | 1_isocortex | 3 | 3.0 | 0 | 1_isocortex | L | F | 1 | 2 | 2 |
| 3 | Cingulate_Cortex_Area_24b_prime | A24bPrime | Left | 1_forebrain | 1_secondary_prosencephalon | 1_isocortex | 2_cingulate_cortex | 1_isocortex | 4 | 4.0 | 0 | 1_isocortex | L | F | 1 | 3 | 5 |
| 4 | Cingulate_Cortex_Area_29a | A29a | Left | 1_forebrain | 1_secondary_prosencephalon | 1_isocortex | 2_cingulate_cortex | 1_isocortex | 5 | 5.0 | 0 | 1_isocortex | L | F | 0 | 0 | 0 |
## Plot to make sure nothing is wrong
for l in range(3):
fig, ax = plt.subplots(
ncols=3,
figsize=(20, 10),
#constrained_layout=True,
dpi=300,
gridspec_kw=dict(width_ratios=[1, 1, 1])
)
for (i, genotype) in enumerate(cor_dat.keys()):
gp.plot.adjplot(
cor_dat[genotype],
ax=ax[i],
vmin=-1,
vmax=1,
meta=meta,
group=[f'Cluster_{l+1}'],
)
ax[i].set_title(f"{genotype}", pad=90, size=30)
#fig.savefig(f"./figures/2022-02-02-multigraph-clustering-level-{l + 1}.png", bbox_inches='tight')
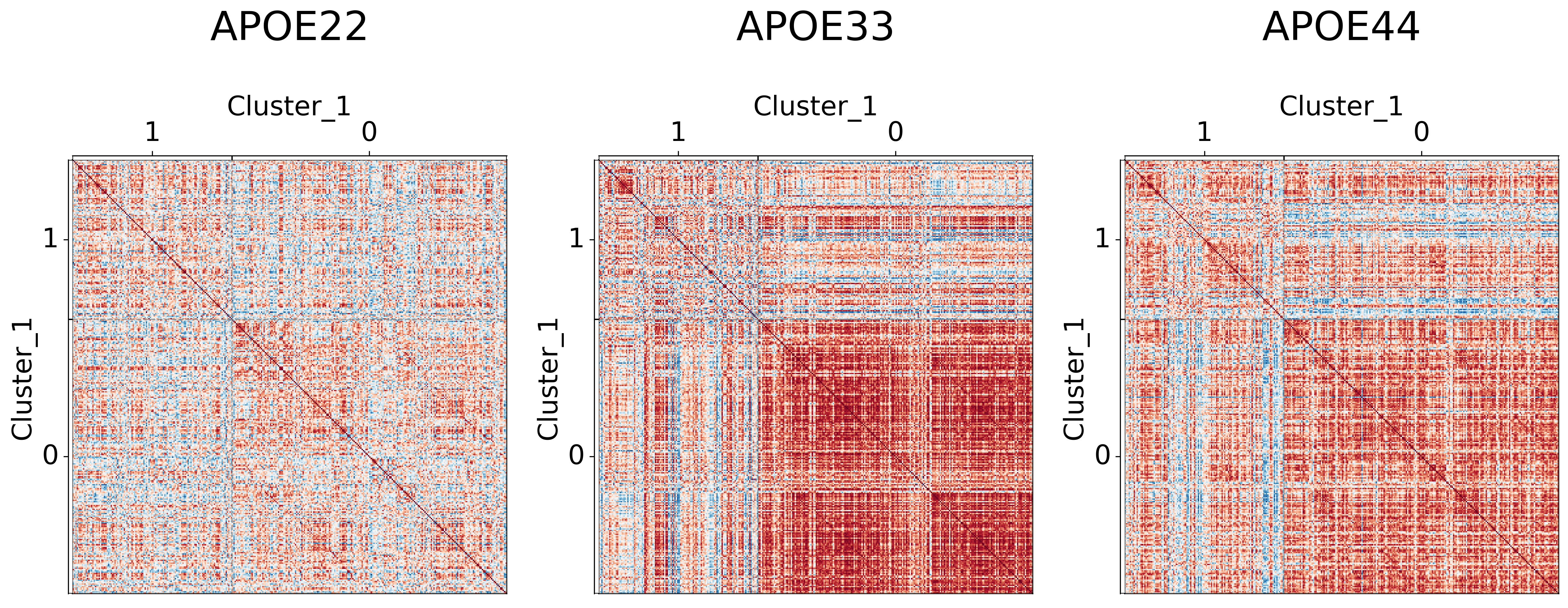
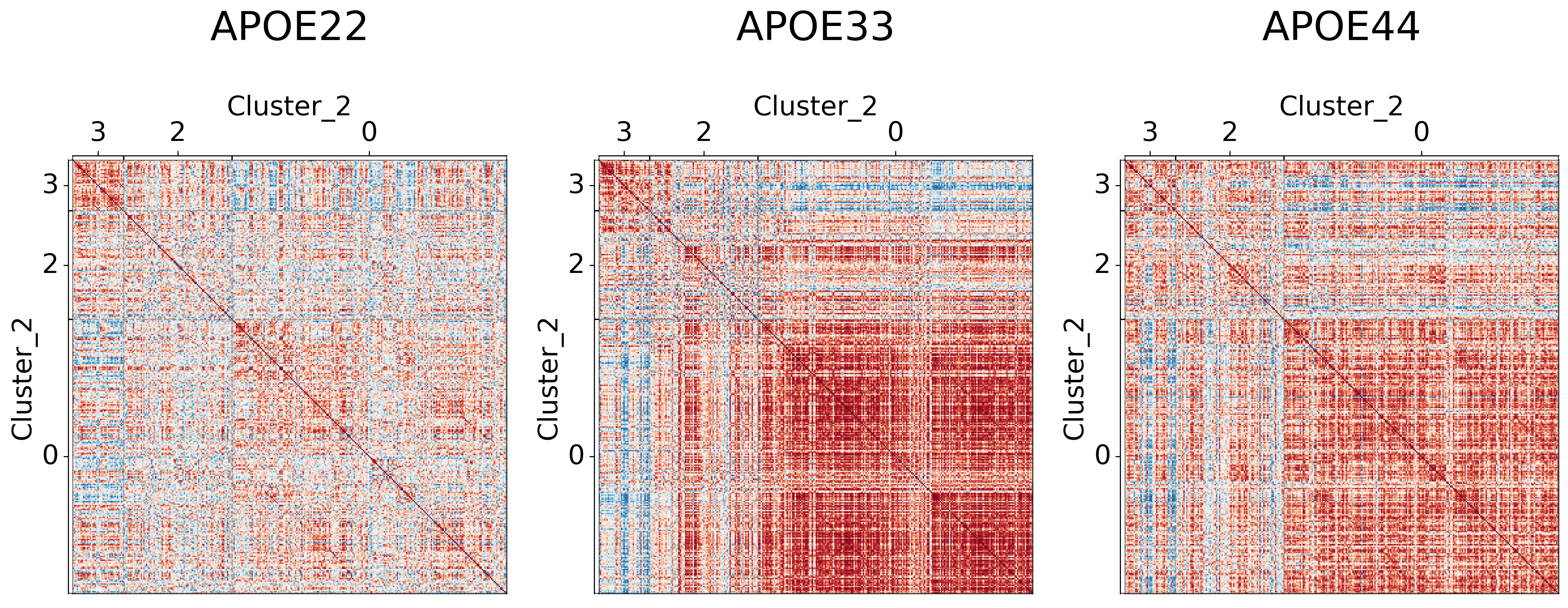
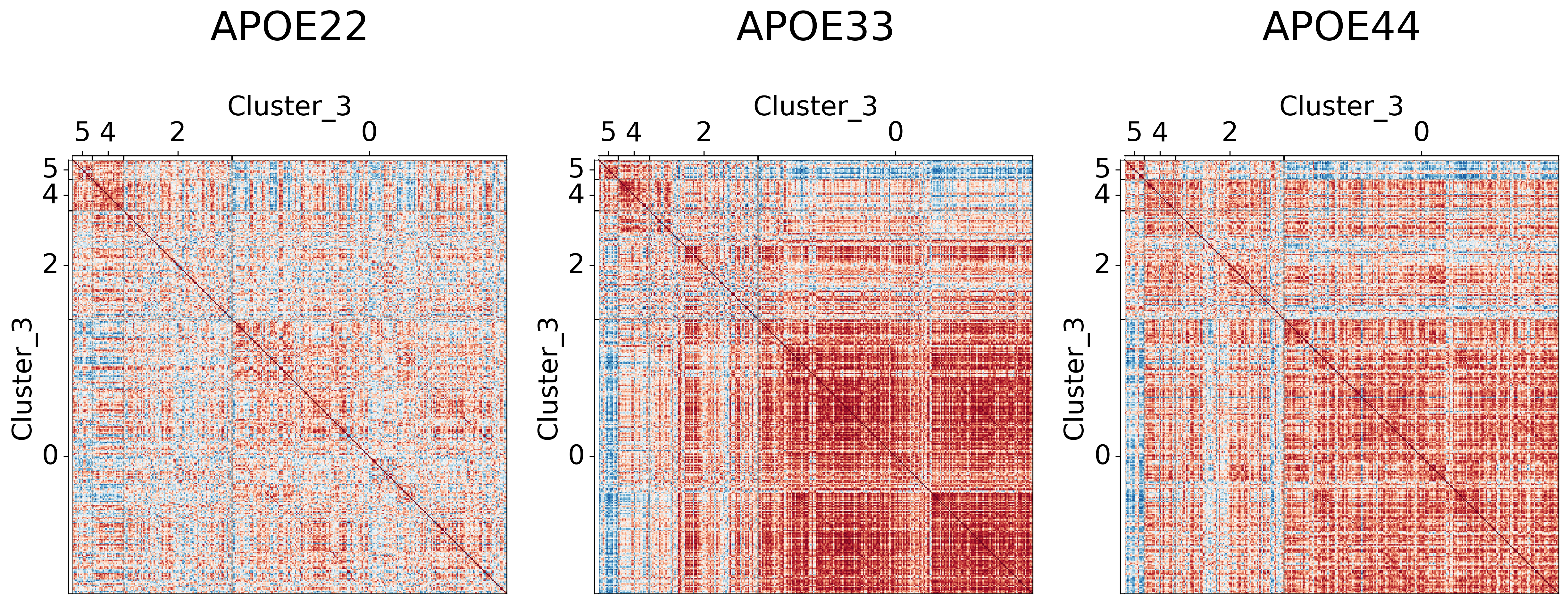
It seems like cluster structures are predominantly driven by the large correlations in the APOE3 genotype
Testing for significantly different clusters at various levels#
Again, the clusters tell us which regions should be grouped together. Hence, each cluster forms our subgraph. For each subgraph, we test whether the distribution of the latent positions (aka the embeddings) are significantly different. Specifically we test
using a 3-sample distance correlation. We correct for multiple hypothesis testing via Holm-Bonferroni correction.
def holm_bonferroni(pvals, alpha=0.05):
sorted_pvals = np.sort(pvals)
m = len(pvals)
corrected_alpha = alpha / m
for i in range(m):
if sorted_pvals[i] < corrected_alpha:
if (m + 1 - (i + 2)) == 0:
return alpha / m
corrected_alpha = alpha / (m + 1 - (i + 2))
else:
break
return corrected_alpha
res = []
for level in range(cluster_labels.shape[1]):
level_labels = cluster_labels[:, level]
uniques = np.unique(level_labels)
for cluster in uniques:
idx = (level_labels == cluster)
data = [Xhat[idx] for Xhat in Xhats]
ksample = hyppo.ksample.KSample("dcorr")
stat, pval = ksample.test(*data)
res.append((level, cluster, pval))
ksample_df = pd.DataFrame(res, columns=["Cluster_level", "Cluster_id", "pval"])
corrected_alpha = holm_bonferroni(ksample_df.pval)
ksample_df["significant"] = ksample_df.pval < corrected_alpha
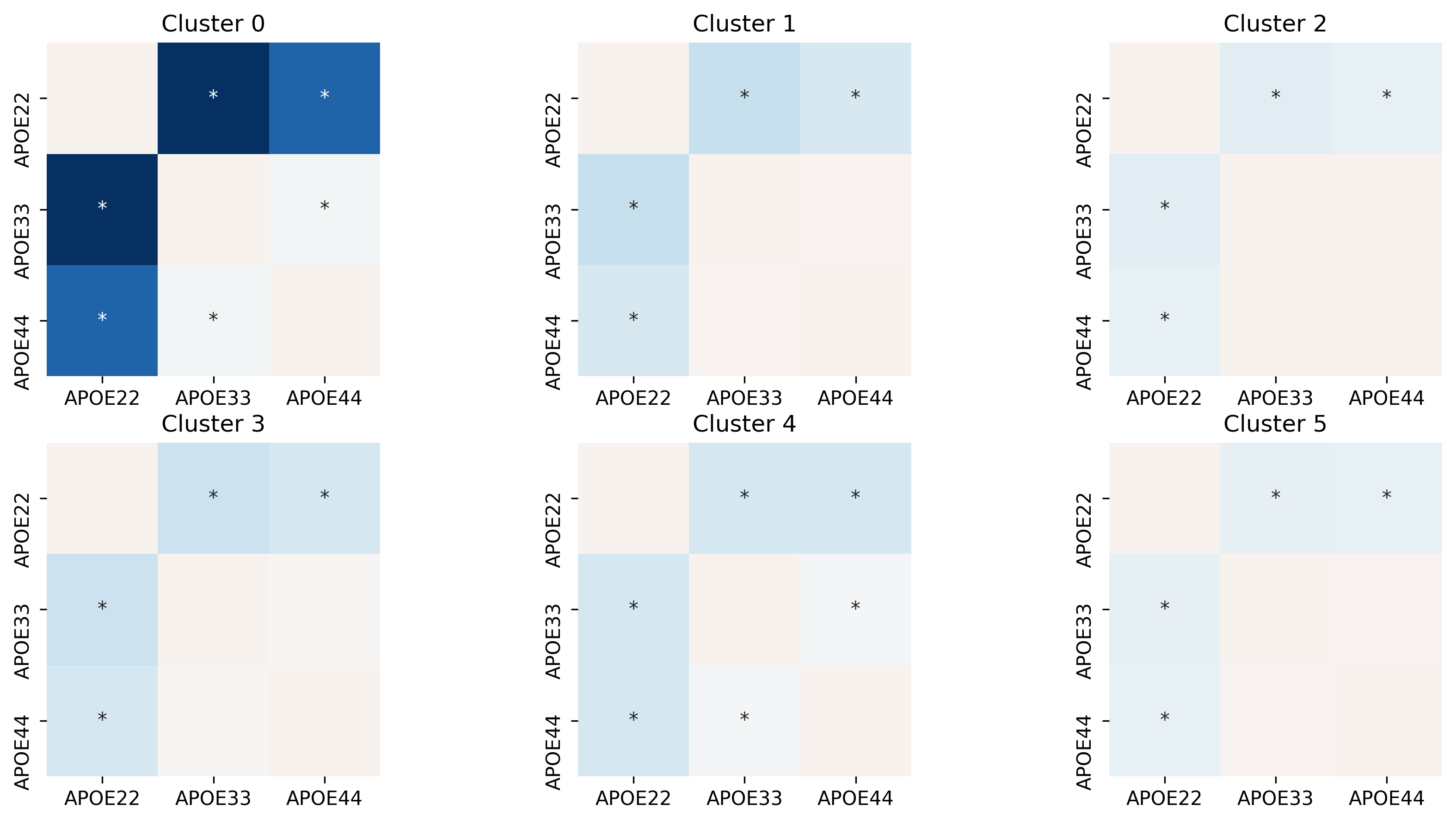
Ksample testing results#
We see that most clusters are significantly different.
ksample_df
| Cluster_level | Cluster_id | pval | significant | |
|---|---|---|---|---|
| 0 | 0 | 0 | 1.774613e-51 | True |
| 1 | 0 | 1 | 1.149383e-12 | True |
| 2 | 1 | 0 | 1.774613e-51 | True |
| 3 | 1 | 2 | 2.072957e-07 | True |
| 4 | 1 | 3 | 1.937743e-12 | True |
| 5 | 2 | 0 | 1.774613e-51 | True |
| 6 | 2 | 2 | 2.072957e-07 | True |
| 7 | 2 | 4 | 1.825486e-12 | True |
| 8 | 2 | 5 | 5.191775e-07 | True |
Testing for differences in pairwise subgraph covariances#
The test above tells us whether a pair, or all, genotypes are different. For example, a significant result may mean that APOE2 and APOE3 are different. However, it is also possible that APOE2, APOE3, and APOE4 are all different. To figure out which pairs are different, we do a post-hoc pairwise test. That is, for each cluster (aka subgraph), we test whether each pair of genotypes are different. Specifically,
where \(i, j\) denotes a particular genotype. Again, we use distance correlation with Holm-Bonferroni correction.
genotypes = ['APOE22', 'APOE33', 'APOE44']
ksample = hyppo.ksample.KSample("dcorr")
res = []
cluster_df = ksample_df[ksample_df.significant == True]
for _, row in cluster_df.iterrows():
cluster_id = row.Cluster_id
idx = cluster_labels[:, row.Cluster_level] == cluster_id
cluster_Xhat = [Xhat[idx] for Xhat in Xhats]
for i, g in enumerate(genotypes):
for j, h in enumerate(genotypes):
if i < j:
continue
if g == h:
#res.append([cluster_id, g, h, 1])
continue
else:
ksample = hyppo.ksample.KSample("dcorr")
_, pval = ksample.test(cluster_Xhat[i], cluster_Xhat[j])
res.append([row.Cluster_level, cluster_id, g, h, pval])
res = pd.DataFrame(res, columns = ["cluster_level", "cluster_id", "Genotype_1", "Genotype_2", "pval"])
res["log(pval)"] = np.log(res.pval)
corrected_alpha = holm_bonferroni(res.pval)
res["significant"] = res.pval < corrected_alpha
from scipy.spatial.distance import squareform
dfs = [res[0:6], res[9:15], res[-6:]]
res = pd.concat(dfs, axis=0)
fig, ax = plt.subplots(ncols = 3, nrows = 2, figsize=(14, 7), dpi=300)
ax = ax.ravel()
for i, cluster_id in enumerate(np.unique(res.cluster_id)):
tmp = res[res.cluster_id == cluster_id]
data = squareform(tmp["log(pval)"])
sig_labels = squareform(["*" if sig is True else "" for sig in tmp.significant])
sns.heatmap(
data,
annot=sig_labels,
fmt = '',
vmax=0,
vmin=np.min(res["log(pval)"]),
xticklabels=genotypes,
yticklabels=genotypes,
center = np.log(corrected_alpha),
cmap="RdBu_r", square = True, ax = ax[i], cbar=False)
ax[i].set_title(f"Cluster {cluster_id}")
#out = pd.concat([node_labels, cluster_label_df], axis=1)
#out.to_csv("./out.csv")
Jesús Arroyo, Avanti Athreya, Joshua Cape, Guodong Chen, Carey E. Priebe, and Joshua T. Vogelstein. Inference for multiple heterogeneous networks with a common invariant subspace. Journal of Machine Learning Research, 22(142):1–49, 2021. URL: http://jmlr.org/papers/v22/19-558.html.
Thomas L Athey, Tingshan Liu, Benjamin D Pedigo, and Joshua T Vogelstein. Autogmm: automatic and hierarchical gaussian mixture modeling in python. arXiv preprint arXiv:1909.02688, 2019.
Vince Lyzinski, Daniel L Sussman, Minh Tang, Avanti Athreya, and Carey E Priebe. Perfect clustering for stochastic blockmodel graphs via adjacency spectral embedding. Electronic journal of statistics, 8(2):2905–2922, 2014.
Carey E Priebe, Youngser Park, Joshua T Vogelstein, John M Conroy, Vince Lyzinski, Minh Tang, Avanti Athreya, Joshua Cape, and Eric Bridgeford. On a two-truths phenomenon in spectral graph clustering. Proceedings of the National Academy of Sciences, 116(13):5995–6000, 2019.