Brain Volume Analysis#
import pandas as pd
import graspologic as gp
import seaborn as sns
import hyppo
import matplotlib.pyplot as plt
import numpy as np
import plotnine as p9
## Read the data
key = pd.read_csv('../data/processed/key.csv')
data = pd.read_csv('../data/processed/mouses-volumes.csv')
data.set_index(key.DWI, inplace=True)
genotypes = ['APOE22', 'APOE33', 'APOE44']
gen_animals = {genotype: None for genotype in genotypes}
for genotype in genotypes:
gen_animals[genotype] = key.loc[key['Genotype'] == genotype]['DWI'].tolist()
vol_dat = {genotype: [] for genotype in genotypes}
for genotype in genotypes:
vol_dat[genotype] = data.loc[gen_animals[genotype]].to_numpy()[:,1:]
## Compute correlations
cor_dat = {genotype: gp.utils.symmetrize(np.corrcoef(dat, rowvar=False)) for (genotype, dat) in vol_dat.items()}
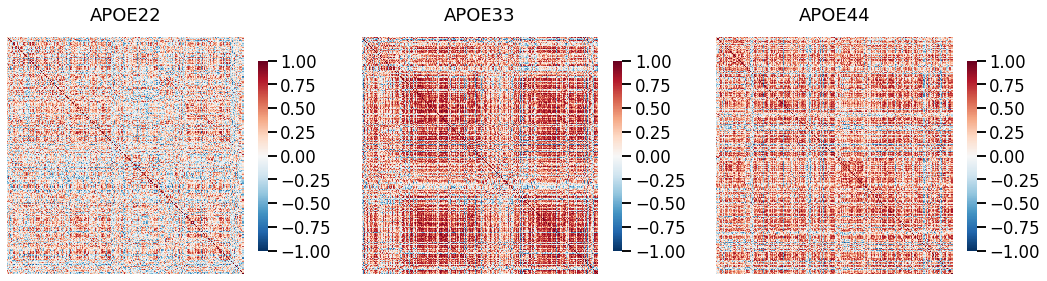
## Plot to make sure nothing is wrong
fig, ax = plt.subplots(1, 3, figsize=(18, 5))
for (i, genotype) in enumerate(cor_dat.keys()):
gp.plot.heatmap(cor_dat[genotype], title=genotype, ax=ax[i], vmin=-1, vmax=1)
Testing whether group volumes are significantly different#
We compute 3-sample distance correlation to see if there is any differences in the 3 groups. Specifically, we test whether the distribution of brain volumes from at least two genotypes are different.
ksample = hyppo.ksample.KSample("dcorr")
stat, pval = ksample.test(*[dat for (genotype, dat) in vol_dat.items()])
print(pval)
0.006007509501547491
We see significant difference among the three genotypes
Testing for most significantly different regions#
Since we detect difference in at least two of the genotypes using the whole brain volumes, we repeat the procedure on each of the regions.
region_pvals = {reg: None for reg in data.columns[1:]}
for i, reg in enumerate(data.columns[1:]):
region_vols = [dat[:, i] for (genotype, dat) in vol_dat.items()]
stat, pval = ksample.test(*region_vols)
region_pvals[reg] = pval
sorted_pvals = dict(sorted(region_pvals.items(), key=lambda item: item[1]))
pd.DataFrame(list(sorted_pvals.items())[:10], columns = ['Region', 'P-value'])
| Region | P-value | |
|---|---|---|
| 0 | ml.1 | 0.000590 |
| 1 | DTT | 0.000763 |
| 2 | PnC | 0.001241 |
| 3 | Cb_1_10.1 | 0.001744 |
| 4 | PnRt | 0.002679 |
| 5 | Vll.1 | 0.003626 |
| 6 | ml | 0.004092 |
| 7 | DTT.1 | 0.004172 |
| 8 | SC.1 | 0.004264 |
| 9 | ParB | 0.005457 |
Testing for differences in the brain volume covariance#
Using similar procedure as above, we test whether the covariances are different among the genotypes. Prior to the 3-sample test, we use Omnibus embedding on the covariance matrices to obtain latent positions, which we then test for difference in distributions.
omni = gp.embed.OmnibusEmbed()
omni.fit([dat for (_, dat) in cor_dat.items()])
Xhats = omni.latent_left_
print(Xhats.shape)
(3, 332, 4)
ksample = hyppo.ksample.KSample("dcorr")
_, pval = ksample.test(*Xhats)
print(pval)
8.254124554923335e-26
We again see a significant difference in distributions
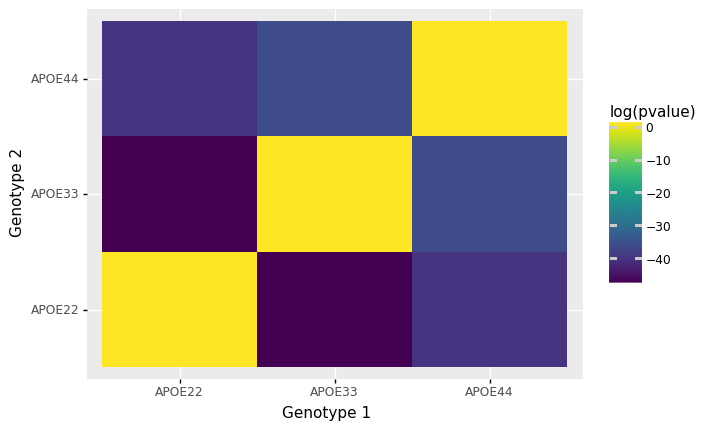
Testing for differences in pairwise covariances#
We then test for whether the distribution of each pair of genotypes are different.
genotypes = ['APOE22', 'APOE33', 'APOE44']
ksample = hyppo.ksample.KSample("dcorr")
res = []
for i, g in enumerate(genotypes):
for j, h in enumerate(genotypes):
if g == h:
res.append([g, h, 1])
else:
_, pval = ksample.test(Xhats[i], Xhats[j])
res.append([g, h, pval])
pairwise_df = pd.DataFrame(res, columns=["Genotype 1", "Genotype 2", "pvalue"])
pairwise_df["Genotype 1"] = pairwise_df["Genotype 1"].astype('category')
pairwise_df["Genotype 2"] = pairwise_df["Genotype 2"].astype('category')
pairwise_df["log(pvalue)"] = np.log(pairwise_df["pvalue"])
pairwise_df
| Genotype 1 | Genotype 2 | pvalue | log(pvalue) | |
|---|---|---|---|---|
| 0 | APOE22 | APOE22 | 1.000000e+00 | 0.000000 |
| 1 | APOE22 | APOE33 | 9.932969e-21 | -46.058427 |
| 2 | APOE22 | APOE44 | 1.014474e-17 | -39.129576 |
| 3 | APOE33 | APOE22 | 9.932969e-21 | -46.058427 |
| 4 | APOE33 | APOE33 | 1.000000e+00 | 0.000000 |
| 5 | APOE33 | APOE44 | 3.573940e-16 | -35.567693 |
| 6 | APOE44 | APOE22 | 1.014474e-17 | -39.129576 |
| 7 | APOE44 | APOE33 | 3.573940e-16 | -35.567693 |
| 8 | APOE44 | APOE44 | 1.000000e+00 | 0.000000 |
plot = (p9.ggplot(p9.aes(x="Genotype 1", y="Genotype 2", fill="log(pvalue)")) +
p9.geom_tile(pairwise_df))
plot
<ggplot: (8766045953917)>